Нейрофиброматоз 1 типа противопоказания

Нейрофиброматоз 1 типа — методы диагностики, лечения по Европейским рекомендацияма) Синоним. Нейрофиброматоз 1 типа (НФ1) также известен как периферический нейрофиброматоз (НФ) или болезнь фон Реклингхаузена. б) Эпидемиология. Наиболее распространенная форма нейрофиброматоза (НФ) составляет около 96% случаев с заболеваемостью I на 4000 живорожденных. Гендерное и расовое преобладание не описано. в) Этиология. Нейрофиброматоз 1 типа (НФ1) наследуется по аутосомно-доминантному типу почти с полной пенетрантностью, но варьирующей экспрессией. Тем не менее, положительный семейный анамнез отмечается почти у 50% пациентов, остальные 50% случаев, как полагают, представляют собой новые мутации. Участвует ген супрессоров опухолей, Ген расположен в перицентрической области на длинном плече 17-й хромосомы и имеет чрезвычайно высокую скорость спонтанной мутации более чем с 200 вариантами, известными на настоящий момент. Продукт гена, нейрофибромин, представляет собой большой цитоплазматический белок, существующий в нескольких формах в различных тканях; он, как полагают, взаимодействует с внутриклеточными цитоплазматическими микротрубочками. г) Диагностика и лечение нейрофиброматоза 1 типа. Из-за отсутствия общепринятого генетического теста диагноз основан на клинических проявлениях. Они не всегда присутствуют с рождения, проявляясь в разные периоды жизни. 1. Кожные проявления: 2. Глазные проявления: 3. Опорно-двигательные нарушения: 4. Неврологические проявления: — Периферические нейрофибромы: множественные кожные, подкожные и периферические поражения, часто распространяются в торакоабдоминальной области или вокруг соска. Гистологически доброкачественные (злокачественные преобразования редки и могут быть представлены в количестве от нескольких до нескольких тысяч). В основном они представляют собой косметическую проблему с возможностью определенной хирургической коррекции по желанию пациента. — Плексиформные нейрофибромы: часто поражают большое количество периферических нервов или часть симпатической цепочки с потенциальным обезображиванием (гемигипертрофия руки/ноги) или нарушением функции вовлеченных областей. В целом доброкачественные, но злокачественное преобразование происходит (злокачественная оболочки периферических нервов опухоль: MPNST) в 6% случаев. Для бессимптомных поражений предпочтительна тактика выжидания и наблюдения; результаты лечения с 13-цис-ретиноевой кислотой и интерфероном альфа-2а находятся в стадии оценки. — Параспинальные нейрофибромы: наиболее распространенные опухоли, поражающие позвоночник у пациентов с нейрофиброматозом 1 типа (НФ1). Параспинальные нейрофибромы, как правило, возникают из спинальных корешков в шейном и поясничном отделах, они могут врасти в позвоночный канал через межпозвонковые отверстия, в результате чего формируются гантелевидные поражения. Хирургическая резекция показана во всех симптоматических случаях и по рентгенологическим критериям (масс-эффект) для шейно-грудной области (риск вторичной миелопатии в связи со сдавлением спинного мозга). — Глиомы зрительных путей: наиболее распространенное проявление в центральной нервной системе при нейрофиброматозе 1 типа (НФ1), поражает примерно 15% пациентов. Эта опухоль обычно встречается в детском возрасте с наибольшим риском возникновения в течение первых шести лет жизни; женщины страдают вдвое чаще, чем мужчины. Развитие возможно в любой точке зрительного пути, но чаще всего встречаются прехиазмальные поражения. Приблизительно половина всех оптических глиом протекает бессимптомно, и большинство из них, в том числе симптоматические, редко прогрессируют. Глазные признаки являются наиболее частыми клиническими проявлениями; у 39% детей с поражениями хиазмы выявляется гипоталамо-гипофизарная дисфункция. д) Рекомендации: Хирургическое лечение ограничено определенными показаниями: у детей с внутриглазничным и прехиазмальным поражениями и закрывающимися глазами (или проптоз глаза с тяжелыми формами нарушения зрения) операция может быть выполнена в косметических целях и для предотвращения распространение опухоли на хиазму. При хиазмальных опухолях вмешательство может потребоваться для уменьшения объема больших опухолей, особенно с кистозным компонентом. У детей старше трех лет лучевая терапия является вариантом лечения, почти 80% случаев дают стабилизацию/уменьшение опухоли. Тем не менее, и в таких случаях при лучевой терапии существует риск нейрокогнигивных и нейроэндокринных последствий, помимо вызываемых облучением злокачественных новообразований. За последние 15 лет ряд клинических исследований показал, что прогрессирование опухоли может быть приостановлено при использовании одно- и мультиагентной химиотерапии, и, следовательно, химиотерапия в настоящее время предлагается в качест ве терапии первой линии, особенно у более молодых пациентов (моложе трех лет), для которых лучевая терапия противопоказана. — Аномалии ликворных пространств: некоторое расширение желудочков относительно часто выявляется при нейрофиброматозе 1 типа (НФ1), но его роль в патогенезе задержки психомоторного развития спорна. Действительно, в большинстве случаев расширение желудочковой системы не связано с повышением внутричерепного давления и не является прогрессирующим. Это относится и к расширению субарахноидальных влагалищ корешков спинного мозга. Лишь в исключительных случаях эти менингеальные растяжения могут стать симптоматическими при сжатии или деформации корешков спинного мозга, следовательно, будет необходимо лечение (как правило, шунтирование). — Другие внутричерепные опухоли: глиомы полушарий встречаются менее чем в 0,5%, а глиомы задней ямки менее, чем в 1% случаев нейрофиброматоза 1 типа (НФ1). Большинство этих опухолей не имеет тенденции к росту. По этой причине, как и для поражений зрительных путей, последовательный мониторинг (клинический и рентгенологический) целесообразен в бессимптомных случаях, а оперативное вмешательство показано для пациентов с симптомами и/или опухолями с тенденцией к росту. Неопознанные яркие включения при МРТ — частое явление у больных с НФ1. Они представлены областями спонтанной гиперинтенсивности на Т2-взвешенных изображениях, чаще всего с участием базальных ганглиев, в мозжечке, в стволе головного мозга и подкорковом белом веществе. Предполагается, что они представляют собой участки миелинопатии, которые, как правило, разрешаются спонтанно. — Неврологическая задержка развития: 30-65% пациентов с нейрофиброматозом 1 типа (НФ1) проявляют некоторую степень неспособности к обучению, речевые навыки сохраняются лучше, чем визуальнопространственные. е) Прогноз. При самом лучшем медицинском обслуживании продолжительности жизни при нейрофиброматозе 1 типа (НФ1) остается примерно на 15 лет меньше, чем у населения в целом. Озлокачествление является одной из основных причин снижения ожидаемой продолжительности жизни у взрослых с НФ1, а общий риск злокачественных осложнений составляет примерно 3-15%.
— Также рекомендуем «Нейрофиброматоз 2 типа — методы диагностики, лечения по Европейским рекомендациям» Оглавление темы «Нейрокожные синдромы.»:
|

Общие сведения
Нейрофиброматоз — это группа наследственных относительно редких заболеваний, проявляющихся множеством симптомов и фенотипической вариабельностью. Наиболее часто нейрофиброматоз (НФ) проявляется характерными патологическими изменениями — развитием опухолей преимущественно эктодермального происхождения с поражением кожного покрова, центральной нервной системы, наличием специфических пигментных пятен на коже по цвету «кофе с молоком», аномалиями развития костного скелета и рядом других проявлений.
Код нейрофиброматоз по мкб-10: Q85.0 (нейрофиброматоз незлокачественный). Нейрофиброматоз относится к наиболее распространенной форме наследственной моногенной патологии, частота встречаемости которого варьирует в пределах 1:2000 — 1:40000. Характерен аутосомно-доминантный тип наследования заболевания с частотой проявления (пенетрантностью) приближающейся к 100%. Различий в частоте поражения по половому признаку не выявлено.
В целом по клинико-морфологическим проявлениям выделено и описано восемь типов нейрофиброматоза, наиболее значимыми из которых являются:
- Нейрофиброматоз первого типа (НФ 1). Около 90% всех нейрофиброматозов приходится на тип НФ1, который и является наиболее распространённым среди заболеваний этой группы.
- Нейрофиброматоз второго типа (НФ 2). Встречается значительно реже (1:40000), но имеет более агрессивное течение, чем НФ1.
Некоторые авторы считают, что из восьми форм нейрофиброматоза все, кроме НФ2 считаются абортивными и не должны выделяться в качестве самостоятельных нозологических форм.
Нейрофиброматоз 1 типа (синоним «болезнь Реклингаузена»)
Частота встречаемости у новорожденных составляет 1:4000, в русской популяции — 1,28:10 000. Более характерна передача по отцовской линии, чем по материнской. В более половине случаев проявления заболевания минимальны, часто встречаются неполные и моносимптомные формы. Около 80% случаев нейрофиброматоза спорадические и являются результатом новых мутаций. В связи с присущей гену NF1 высокой частоты мутаций, определяющих клиническое развитие нейрофиброматоза, предполагается или чрезвычайно высокая его мутабельность или наличие мутаций в нескольких локусах гена.
Для нейрофиброматоза I типа характерен выраженный полиморфизм клинических проявлений, прогрессирующее течение и частое вовлечение в процесс различных органов и систем с развитием тяжелых осложнений, нередко приводящих к летальному исходу. Болезнь Реклингхаузена (нейрофиброматоз) имеет четко прослеживающуюся тенденцию к медленному прогрессированию. Существует два периода резкого увеличения активности патологического процесса: первый от 5 и до 10 и второй от 35 до 50 лет. При этом, второй период активности в подавляющем числе случаев (около 75%) обусловлен малигнизацией опухолевых новообразований. К провоцирующим факторам, способствующим росту нейрофибром, относятся состояния и возрастные изменения, сопровождающиеся гормональной перестройкой организма: беременность, период полового созревания.
Особенность заболевания является специфическая последовательность манифестации симптоматики нейрофиброматоза 1-го типа в зависимости от возраста пациента, что и обуславливает трудности в его диагностике у детей раннего возраста. Так, нейрофиброматоз у детей первых лет жизни/с рождения может присутствовать только в виде некоторых признаков заболевания, чаще — это крупные пигментные пятна или скелетные дисплазии и плексиформные нейрофибромы, а другие характерные симптомы могут манифестировать значительно позже (к 5-15 годам). А если учесть, что степень их выраженности, течение и скорость прогрессирования патологического процесса варьируют в широких пределах становится понятными затруднения в постановке диагноза, что и способствует его поздней диагностике.
Нейрофиброматоз 2 типа
Нейрофиброматоз II типа встречается независимо от НФ-I типа; его проявления определяются локализацией основного патологического процесса и соответствует симптоматике поражения головного/спинного мозга. В спинном мозге чаще в процесс вовлекаются оболочки спинальных корешков шейного и поясничного отделов в виде Шванном (опухоль доброкачественная из шванновских клеток нервных оболочек), конский хвост (задние корешки); при локализации в головном мозге в процесс могут вовлекаться черепно-мозговые нервы, продолговатый мозг и Варолиев мост. Реже в процесс вовлекаются периферические нервы с развитием эпендимом и менингиом.
Средний возраст манифестации признаков заболевания при НФ 2 составляет 20 лет, а на момент постановки диагноза – около 28 лет. Как правило, НФ 1 начинает проявляться в раннем детстве, преимущественно с кожных симптомов, в то время как НФ 2 проявляется в молодом возрасте, преимущественно с развития глухоты, обусловленной развитием вестибулярных шванном и других симптомов, вторичных относительно спинальных шванном и менингиом.
Патогенез
В основе патогенетических изменений нейрофиброматоза 1 типа лежит высокая частота спонтанных мутаций гена NF1 (17q11.2), кодирующего цитоплазматический белок-онкосупрессор нейрофибромин, который вырабатывается в нервных и специализированных клетках нейроглии. Нейрофибромин содержит в своем составе особый домен, который и реализует супрессорный эффект относительно пролиферации клеток преимущественно нейроэктодермального происхождения за счет инактивации адаптерного белка Ras, который является ингибитором структуры митогенных сигнальных путей. При такого рода генетическом дефекте в хромосомах 17q происходит нарушение динамического равновесия регуляции роста и его смещение в сторону пролиферации и образования доброкачественного опухолевого роста.
Описано более 500 видов мутаций в гене на хромосоме 17q, которые и препятствуют осуществлению регулирующей функции гена NF1 в онкогенезе. Механизм развития клинических проявлений до настоящего времени точно неизвестен. Принято считать, что в основе патологии лежит развитие из нервных оболочек различных опухолей (невриномы, нейрофибромы). Примерно половина случаев являются следствием новых мутаций.
Генетический дефект при нейрофиброматозе 2 типа располагается в 22 хромосоме (22q12) и непосредственно кодирует процесс синтеза белка Мерлина, также являющегося супрессором опухолевого роста. Наибольшее значение он имеет в регулировании пролиферации (размножения) клеток нейроэктодермального происхождения. При мутации синтеза Мерлина в одной хромосоме не проявляется на клеточном уровне, а при симметричной мутации (повреждении) аллельного гена в клетке прекращается синтез Мерлина и соответственно, присущее здоровому организму равновесие в регуляции роста смещается (дрейфует) в сторону пролиферации с развитием процесса доброкачественного опухолевого роста.
Классификация
В основе классификации нейрофиброматоза лежат клинико-морфологические особенности заболевания в соответствии с чем выделяют:
- Периферический нейрофиброматоз (1 тип)— в клинической картине доминируют поражение периферических нервов и кожных покровов.
- Центральный нейрофиброматоз (2 тип) — в патологический процесс вовлекаются преимущественно черепные нервы и спинальные корешки.
Причины
Этиологическим фактором нейрофиброматоза 1 типа являются множественные мутации одного из самых протяженных и сложно организованных генов NF1 (17q11.2), являющихся следствием инактивации преимущественно отцовской аллели. Причиной нейрофиброматоза 2 типа является генетический дефект в 22 хромосоме (22q12).
Симптомы
Нейрофиброматоз Реклингхаузена начинает проявляться в большинстве случаев множественными нейрофибромами, с локализацией по ходу периферических нервов, в виде округлых болезненных узелков, расположенных в толще кожи, различающиеся по своим размерам. Нейрофибромы представлены в виде узелков на/в толще кожи нормальной или синюшно-красной окраски и мягко эластической консистенции.
Характерным признаком, начиная с периода новорожденности/первых лет жизни детей является появление на коже мелких «кофейных пятен», напоминающих веснушки с четкими границами и ровными краями с преимущественной их локализацией на участках тела со складками (в паховой области, подмышечных впадинах, на животе). По мере взросления ребенка наблюдается тенденция к увеличению их численности (от единичных пятен до нескольких сотен) и размеров (от точечных до 2-5 сантиметров в диаметре), они также могут приобретать темную окраску. Следует учитывать, что к диагностически значимым пятнам относятся у детей до пубертата — диаметром более 5 мм и после полового созревания — 15 мм и более. Нейрофиброматоз (фото начальной стадии) приведены ниже.

Мягкие кожные опухоли, как правило, у детей отсутствуют, особенно первых лет жизни и возникают к 10-15 годам. При этом, их количество особенно возрастает в пубертатном периоде. При пальпации они безболезненны, однако при вовлечении в патологический процесс периферических нервов, могут возникают боли, гипестезии. На коже могут присутствовать и другие изменения: участки депигментации, сосудистые пятна, очаговое поседение волос, гипертрихоз. Возможно развитие нейрофибром. Плексиформная нейрофиброма может образовывать гигантские опухоли, состоящие из кожных и подкожных элементов.
Некоторые авторы (В.В. Мордовцева) выделяет четыре варианта клинического течения нейрофиброматоза I типа:
- с наличием незначительных пигментных пятен типа веснушек в сочетании с крупными «кофейными» пятнами при отсутствии нейрофибром или с наличием единичных опухолей;
- с наличием преимущественно крупных пигментных кофейных пятен на коже и небольшим количеством нейрофибром;
- с преобладанием нейрофибром и незначительным количеством пигментных пятен (по типу веснушек/крупных);
- смешанный тип.
Факультативные симптомы нейрофиброматоза проявляются изменениями в костной системе в виде различных деформаций позвоночного столба — сколиозы, лордосколиозы, кифосколиозы, псевдоартрозы, утончение/искривление длинных трубчатых костей, патологических переломов. Нередко развиваются лицевые дисморфии: аномалии глазных щелей, глазной гипертелоризм, деформация ушных раковин, неправильная форма черепа и др. Для пациентов с НФ-I характерны макроцефалия, низкий рост, дисплазия клиновидной кости и затылочной части черепа.
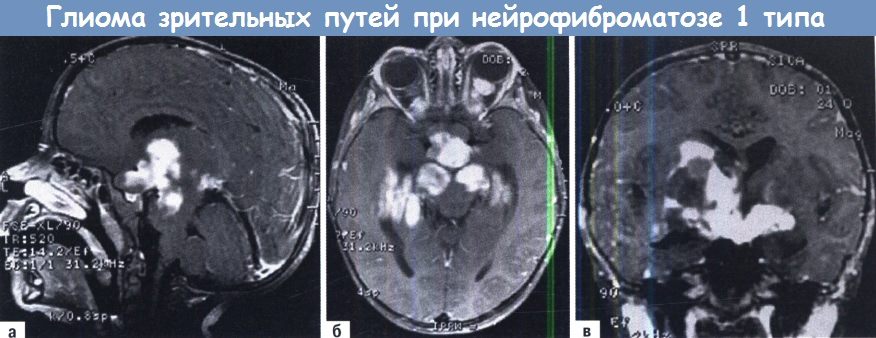
Характерными поражениями нервной системы являются судорожный и гипертензионно-гидроцефальный синдромы, опухоли ЦНС (эпендимома, астроцитома, глиома зрительного нерва/ствола мозга), эпилепсия, что появляется соответствующей симптоматикой. Так, глиома зрительного нерва проявляется нарушением зрения.
Для NF1 характерны дополнительные проявления в виде когнитивных расстройств от легких до выраженных, часто в сочетании с умеренным снижением IQ ребенка, затруднением в освоении чтения, письма, математики и нервно-психической неуравновешенностью; наличие эндокринных расстройств (полового созревания, феохромоцитома, нарушение роста и др.). Со стороны сосудов у пациентов часто регистрируется стеноз почечных артерий с повышением АД, окклюзия артерий, болезнь Мойа-Мойа.
Клиническая манифестация заболевания НФ2 включает развитие двусторонних вестибулярных шванном или шванном других черепных периферических и спинальных нервов при минимальных экстраневральных и кожных симптомах. Двусторонняя шваннома (невринома) слухового нерва относится к основным признакам, который встречается практически в 90% случаев, редко обнаруживается у детей, развиваются преимущественно в позднем подростковом/раннем взрослом возрасте. НФ-II.
Значительно реже (около 10%) случаев встречаются различного рода менингиомы (спинальные, интракраниальные, менингиомы зрительных нервов), глиомы и эпиндимомы. Первым симптомом часто является снижение/потеря слуха, появление в ушах шума, который может сопровождаться атаксией и головокружением. Пятна на коже цвета «кофе с молоком» в большинстве случаев отсутствуют или присутствуют в небольшом количестве. У 50-70% больных нейрофиброматозом 2 типа диагностируются зрительные нарушения (гемартромы, катаракты, ретинобластомы, менингиомы зрительных нервов).
Ниже в таблице приведены характерные проявления нейрофиброматоза 1 и 2 типа.
Анализы и диагностика
Диагноз нейрофиброматоза 1 типа ставится на основе абсолютных и относительных клинических признаков и данных инструментальных методов диагностики:
- КТ/МРТ позвоночника, головного мозга, внутренних органов.
- Рентгенография позвоночника.
- Электрокохлеография.
- Офтальмоскопия.
- Импедансометрия.
- Генетическое обследование и проведение генеалогического анализа.
Лечение
Патогенетическое лечение нейрофиброматоза до настоящего времени не разработано. В большинстве случаев проводится симптоматическая терапия, что и подтверждает специализированный форум. Так, при выраженном болевом синдроме показаны НПВС (Диклофенак, Мелоксикам, Ибупрофен, Нимесулид, Напроксен, Кетопрофен, Пироксикам), неопиодные (Парацетамол, Анальгин, Бутадион, Аспирин и др.) и опиоидные анальгетики (Трамадол, Фентанил, Налбуфин, Тримеперидин), трициклические антидепрессанты (с осторожностью из-за высокого риска судорожного синдрома), Нейронтин (Габапентин), Топирамат. Консервативная терапия предусматривает курсовое назначение препаратов, влияющие на:
- дегрануляцию тканевых базофилов (назначается Кетотифен);
- пролиферацию клеточных элементов (назначаются ретиноиды — Ацитретин, Этретинат, Бексаротен, Тазаротен);
- снижение во внеклеточном матриксе количества гликозаминогликанов (Гиалуронидаза).
Согласно последним рекомендациям, лечение нейрофиброматоза может проводиться с использованием новых перспективных препаратов для таргетной (нацеленной) терапии, воздействующие на определенное звено в патогенетической цепи нейрофиброматоза. Для таргетной терапии используются два вида молекул: мелкие молекулы и моноклональные антитела. К таким препаратам относятся Бевацизумаб (Авастин), Эрлотиниб, Иматиниб, Салиразиб, Лапатиниб (Тайверб).
В случаях малигнизации опухолей проводится химиотерапия и лучевая терапия. При когнитивных нарушениях и плохой обучаемости детей рекомендуется проводить учебный процесс в спецшколах, а также проведение социальной реабилитации взрослых пациентов.
Доктора
Лекарства
- Кетотифен, Этретинат, Тазаротен, Ацитретин, Гиалуронидаза, Диклофенак, Мелоксикам, Ибупрофен, Нимесулид, Напроксен, Кетопрофен, Пироксикам.
- Парацетамол, Анальгин, Бутадион, Аспирин, Трамадол, Фентанил, Налбуфин, Тримеперидин, Нейронтин (Габапентин), Топирамат, Бевацизумаб (Авастин).
- Эрлотиниб, Иматиниб, Салиразиб, Лапатиниб (Тайверб).
Процедуры и операции
При наличии сколиоза и костных деформаций показаны операции ортопедические. При наличии болезненных липом, нейрофибром, папиллом больших размеров или при локализации опухолей в областях частой/постоянной травматизации или являющихся выраженным косметическим дефектом проводятся хирургические операции.
Симптомы нейрофиброматоза у детей
Симптоматика нейрофиброматоза 1 типа у детей аналогична, однако манифестация характерных признаков тесно привязана к возрасту ребенка. Ниже приведена таблица характерных признаков нейрофиброматоза I типа и осложнений у детей в различные возрастные периоды (Википедия).

Наиболее частым проявлением являются «кофейные пятна» на коже ребенка. Ниже приведено фото нейрофиброматоза у детей.

У детей NF1 может быть негрубо нарушена координация движений, снижен мышечный тонус. Также, многие дети имеют больший размер черепа чем у сверстников (превышение более 4 стандартных отклонений). Они могут также отставать в росте. Такие дети чаще малоинициативны и эмоционально ограничены по сравнению со здоровыми сверстниками. Почти 12% детей с NF1, имеют характерный фенотип: антимонголоидный размер глаз, гипертелоризм (увеличенное расстояние между парными органами, например, глазами), низко посаженные уши, стеноз легочной артерии, шейный птеригум. НФ-II у детей обнаруживается крайне редко.
Диета
Специально разработанной диеты при нейрофиброматозах нет.
Профилактика
В основе профилактических мероприятий проведение пренатальной диагностики №Б1, которая является обязательной в случаях наличия больных/бессимптомных/малосимптомных родителей — носителей патологического гена. При признаках роста, увеличения в размерах или изменения плотности и цвета нейрофибром необходима консультация онколога и постоянное наблюдение за больным.
Последствия и осложнения
Осложнения чрезвычайно разнообразны по своему характеру и включают:
- Малигнизацию (злокачественную трансформацию) опухолей.
- Снижение зрения/слепоту.
- Развитие феохромоцитомы.
- Эссенциальную артериальную гипертензию.
- Стеноз почечной артерии.
- Васкулиты/васкулопатии с поражением церебральных/коронных и артерий. Компрессионная невропатия.
- Деформаций позвоночного столба/грудной клетки — сколиозы, лордосколиозы, кифосколиозы, псевдоартрозы.
- Утончение/искривление длинных трубчатых костей, патологические переломы.
- Косметические дефекты.
- Когнитивные нарушения (синдром дефицита внимания).
Прогноз
Прогноз в каждом конкретном случае индивидуальный и во многом определяется тяжестью проявлений заболевания и темпами его прогрессирования. Продолжительность жизни у пациентов с нейрофиброматозом аналогична средней в популяции или уменьшена в среднем на 15% при развитии тяжелых осложнений или малигнизации опухолей. При получении соответствующего уровня образования пациенты при рациональном трудоустройстве могут вести нормальную жизнь. У некоторых больных из-за грубого косметического дефекта развивается депрессивный синдром.
Список источников
- Сергеев А. С. Частота мутаций нейрофиброматоза // Автореферат дисс. канд. биол. наук. М., 1973 (АМН СССР, Ин-т медицинской генетики).
- Шнайдер Н. А. Нейрофиброматоз первого типа: болезнь Реклинхаузена /Н. А. Шнайдер, А. И. Горелов //Сибирское медицинское обозрение. — 2007. — №3 (44). — С. 91-95.
- Козлов А. В. Нейрофиброматоз 2 (НФ2) // Хирургия опухолей основания черепа / Под редакцией А. Н. Коновалова. — М.: ОАО «Можайский полиграфический комбинат», 2004. — С. 169—170. — 372 с
- Уфимцева М.А., Бочкарев Ю.М., Гальперин А.М., Головырина И.Л., Гурковская Е.П. КОЖНЫЕ ПРОЯВЛЕНИЯ БОЛЕЗНИ РЕКЛИНГАУЗЕНА // Современные проблемы науки и образования. – 2016. – № 6.
- Попова А. А Клинико-диагностические аспекты нейрофиброматоза // Университетская медицина Урала – 2016 — №2.